
Orphan Mimics of MS (ORMS)
Our center evaluates and diagnoses adult patients with a wide range of complex neurological white matter and neuroimmune disorders that mimic MS where the cause could be genetic. These diseases include subcortical dementia, ataxia, progressive spastic paraparesis, familial neuroimmunological disorders, and leukodystrophies. We also evaluate familial MS, one of the orphan diseases associated with sporadic MS, and provide care for rare forms of adult white matter disease, including adult-onset leukodystrophies such as Hereditary Leukodystrophy with spheroids and spastic hereditary paraplegia as they can be MS mimickers.
Treatment of these rare diseases can be challenging because there is no standard treatment, even with a clear cause. To provide the best care, it is critical to identify and clinically characterize these patients to coordinate care, treatment, and rehabilitation and advance our understanding with new research for these orphan diseases.
If you are considering a referral for transfer of care or a second opinion, please email us at mssupport@uchc.edu. Before seeing a patient, a comprehensive evaluation of records and clinical imaging will be performed. We will provide ORMS patients with a thorough evaluation and care coordination that includes a neurology assessment, care planning with social work, disability review, physical therapy and rehabilitation (PMR), and a neuropsychological and genetic evaluation, if necessary.
If you would like to discuss your referral with an expert, please email mssupport@uchc.edu.
Resources
Support Groups & Organizations
Clinical Trials
Conditions
Acute disseminated encephalomyelitis is a sudden onset of inflammation of the brain and spinal cord. The sudden inflammation damages myelin, the protective coating on the nerves in the brain and spinal cord, leading to symptoms that might include fever, vision loss, severe weakness, difficulty coordinating movements, and impaired consciousness. Acute disseminated encephalomyelitis is often triggered by infection and can be debilitating. It can be confused with MS, transverse myelitis, and other demyelinating and inflammatory disorders. ADEM is most frequently seen in the pediatric population. We collaborate with experts in pediatric neurology to successfully treat and manage the immediate and long-term consequences of acute disseminated encephalomyelitis.
If you need urgent information regarding a potential patient, please reach out to us.
Acute flaccid myelitis is a rare condition that causes sudden muscle weakness and loss of muscle tone and reflexes. This rare disorder is usually seen 1-4 weeks following a viral flu-like illness and mainly impacts children; however, it can be seen in adults. Additional symptoms include pain, difficulty breathing or swallowing, and problems controlling the bowel or bladder. Acute flaccid myelitis is a debilitating disease and can cause symptoms for months or years following initial onset.
Adult-onset leukodystrophies are a group of genetic diseases where myelin, the protective coating on the nerves in the brain and spinal cord, does not form correctly or is destroyed. There are many different types of leukodystrophies that have each been linked to a specific genetic mutation. Leukodystrophies are progressive diseases, meaning that symptoms worsen over time. Leukodystrophies have various symptoms unique to the particular disease and part of the body affected. Common symptoms include difficulty with movement, balance, coordination, hearing, vision, and cognitive functioning. Leukodystrophies usually present in infancy or childhood but will also present in adulthood on rare occasions. Severity can vary between conditions; however, long-term disease management and care are required. At UConn Health, we have extensive experience treating and managing adult-onset leukodystrophies.
Aicardi-Goutières syndrome is a rare genetic disorder where the myelin, the protective coating on the nerves in the brain and spinal cord, is damaged, and calcium deposits in the brain lead to severe intellectual and physical disability. Patients often present with this disease during infancy or early childhood and typically do not survive past childhood. However, in some milder cases, patients can survive into adulthood. Patients who survive into adulthood might have symptoms that include muscle weakness and stiffness, involuntary muscle movements, developmental delays, and moderate to severe cognitive disability. Several genes cause Aicardi-Goutières syndrome, all inherited in an autosomal recessive pattern. Currently, the only treatment for this disease is supportive therapy and managing the symptoms.
Alexander disease is a rare disorder where there is damage to myelin, the protective coating on nerves in the brain and spinal cord, and abnormal protein deposits throughout the brain. This disease is caused by mutations in a gene called GFAP, which is critical for brain structural support. There are 4 different types of this disease: neonatal, infantile, juvenile, and adult forms, with adult-onset being the least severe and the rarest. For juvenile and adult-onset Alexander disease, symptoms can include impaired coordination, muscle stiffness, trouble speaking, vomiting, and cognitive decline. There is no current cure, and the prognosis for Alexander disease is poor, although the severity can vary. Symptom management and supportive care are currently the only treatment options.
Amyloidosis is a rare disease caused by the build-up of an abnormal protein called amyloid throughout the body. Amyloid protein can accumulate in many different organs, making it difficult to diagnose due to a variable clinical presentation. The most commonly involved organ systems affected by amyloidosis include the kidney, heart, liver, spleen, digestive tract, and brain. Treatment options vary based on the type of amyloidosis and clinical disease severity of the individual. Physicians personalize treatments based on each patient, which often include supportive therapy in addition to the specific treatment. Amyloidosis is a complex and challenging disease to treat and manage. Therefore, individuals are recommended to seek out amyloidosis treatment centers for specialized care. Here at UConn Health, we collaborate with experts to treat and manage the care for patients with amyloidosis.
Behcet’s syndrome is a rare inflammatory disease that causes blood vessel inflammation in many different organs in the body, including the brain, eyes, mouth, skin, lungs, joints, genitals, and gastrointestinal tract. This disease has a variety of symptoms due to inflammation affecting blood vessels of almost all sizes and types. Patients most commonly present with painful canker sores in the mouth but can also develop sores and ulcers on their genitals and in their digestive tract. Patients can also experience eye pain, visual changes, pustules on their skin, joint swelling and pain, headaches, impaired muscle movement, stroke, and difficulty breathing. Every patient with Behcet’s syndrome has a unique presentation based on the organ systems affected, which makes diagnosing Behcet’s disease challenging. Treatments aim to alleviate a patient’s specific symptoms.
CADASIL is a rare genetic disease in which the small blood vessels in the brain become thickened, resulting in reduced blood flow to parts of the brain. This disease is caused by a mutation to a gene called NOTCH3, which is necessary for creating a protein that is critical for the structure of small arteries. Without NOTCH3, the small arteries become damaged over time, leading to symptoms that include migraines, multiple strokes, seizures, psychiatric changes in behavior and personality, and cognitive decline. Individuals with this disease could start to develop symptoms as early as their mid-30s and should receive genetic counseling due to the inheritance pattern of this disease. This disease requires long-term medical management that aims to reduce disability and the severity of the disease. There are currently no treatments for this disease; however, reducing vascular risk factors is necessary for appropriate disease management.
CARASIL is a rare genetic disease where small blood vessels in the brain become damaged, reducing blood flow to parts of the brain. This disease is caused by a mutation in the gene HRTA1 and is inherited in an autosomal recessive pattern. Patients with this disease may experience trouble walking, recurring strokes, mood changes, cognitive decline, difficulty chewing and swallowing, premature hair loss, and back pain that can start as early as 30 years old and progress rapidly. There is no cure for CARASIL, so treatments focus on alleviating the symptoms and providing supportive care.
CTX is a rare disease where individuals can’t break down certain fats, and these fats accumulate throughout the body, most commonly in the brain and tendons. This accumulation can cause many symptoms, including loss of sensation in the arms and legs, seizures, cognitive decline, tremor, difficulty talking, multiple bone fractures, and personality changes. The severity of the symptoms varies with the age of onset and which organ systems have fatty deposits forming. CTX is an autosomal recessive disease, meaning an individual gets a mutated copy of the CYP27A1 gene from both parents. This disease has a specific treatment that has shown to stop disease progression and prevent further symptoms making early diagnosis and treatment of CTX crucial to reduce long-term disease complications.
CLIPPERS is a rare disease that causes progressive inflammation of the brain and spinal cord. Patients will develop symptoms, including trouble walking, double vision, difficulty speaking, and cognitive impairment. The severity increases slowly over weeks and months. Although the cause of CLIPPERS is unknown, steroid treatment can cause significant improvement within a couple of weeks, depending on symptom severity. CLIPPERS can be challenging to diagnose and often presents similarly to other white matter diseases.
Multiple sclerosis is a neurological disease where the body’s immune system attacks myelin, the protective coating on the nerves in the brain and spinal cord. Many patients initially present with sudden, painful vision loss and can develop a wide variety and severity of symptoms based on what parts of their brain and spinal cord are impacted. The most common symptoms include fatigue, trouble walking, bowel or bladder problems, and difficulty with memory. Most cases of multiple sclerosis have no known genetic link. However, a small subset of patients has a family history of multiple sclerosis. This is called familial multiple sclerosis, and these patients tend to present with symptoms at a younger age than patients with sporadic multiple sclerosis. There are currently no cures for multiple sclerosis, so disease management is critical to help slow disease progression. We have extensive experience treating and managing familial multiple sclerosis here at UConn Health.
HDLS is a rare disease where myelin, the protective coating on nerves in the brain and spinal cord, becomes damaged, leading to cognitive and motor decline. This disease is caused by mutations in a gene called CSF1R and inherited in an autosomal dominant pattern, meaning one copy of the mutated gene is enough to cause illness. Symptoms such as personality changes, memory loss, seizures, trouble walking, tremors, muscle stiffness, and paralysis often progress rapidly. The symptoms are thought to occur due to demyelination and axonal swellings called spheroids that are characteristic of this disease. There is no cure, and disease management is essential to alleviate symptoms of the disease.
HAM/TSP is a rare progressive neurological disease that can occur in individuals infected with human T-lymphotropic virus, type I (HTLV-I). Patients can experience various symptoms, including leg weakness, stiff muscles, muscle contractions, exaggerated reflexes, urinary incontinence, and sensations of mild prickling or burning on the skin. Although the onset is slow, it is progressive. There is no cure for this disease, but treatments can help manage disease progression and symptom severity.
Myelin oligodendrocyte glycoprotein associated disease (MOG-AD) is an inflammatory disease where the immune system starts to attack myelin oligodendrocyte glycoprotein (MOG), a component of myelin. Myelin is the protective coating on the nerves in the brain and spinal cord and is necessary for proper nerve function. Individuals with MOG-AD often present with recurrent cases of other demyelinating diseases, including optic neuritis, transverse myelitis, and acute disseminated encephalomyelitis. MOG-AD is commonly confused with multiple sclerosis due to the typical initial onset of optic neuritis.
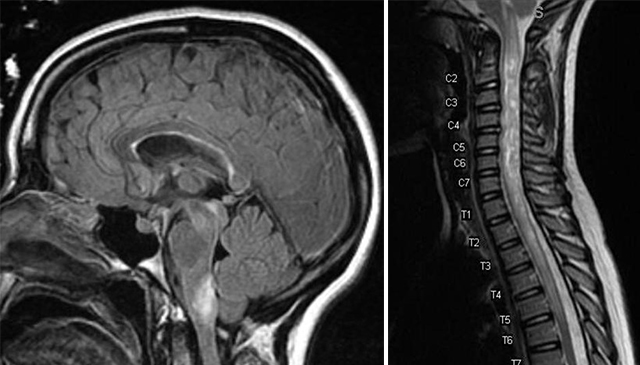 Neuromyelitis optica, also known as Devic’s disease, is a debilitating disease where the immune system attacks the optic nerve and spinal cord resulting in inflammation. Because the individuals experience symptoms related to the part of the optic nerve or spinal cord affected, patients may have a previous diagnosis of optic neuritis, transverse myelitis, or multiple sclerosis. Symptoms can include sudden, painful vision loss, weakness, sensory alterations, or dysfunction of their autonomic nervous system responsible for regulating breathing, digestion, reflexes, and more involuntary bodily functions. Symptomatic episodes will be separated by periods of no symptoms, which might last weeks, months, or even years. There are currently both short-term and long-term treatment options that can help with a current attack and prevent future symptomatic episodes.
Neuromyelitis optica, also known as Devic’s disease, is a debilitating disease where the immune system attacks the optic nerve and spinal cord resulting in inflammation. Because the individuals experience symptoms related to the part of the optic nerve or spinal cord affected, patients may have a previous diagnosis of optic neuritis, transverse myelitis, or multiple sclerosis. Symptoms can include sudden, painful vision loss, weakness, sensory alterations, or dysfunction of their autonomic nervous system responsible for regulating breathing, digestion, reflexes, and more involuntary bodily functions. Symptomatic episodes will be separated by periods of no symptoms, which might last weeks, months, or even years. There are currently both short-term and long-term treatment options that can help with a current attack and prevent future symptomatic episodes.
Neurosarcoidosis is a subtype of sarcoidosis that impacts the brain and spinal cord. Sarcoidosis is an inflammatory disease where granulomas, abnormal clumps of cells, accumulate in organs, resulting in organ damage. In neurosarcoidosis, the granulomas mainly accumulate in the brain, resulting in symptoms such as vision and hearing impairments, headaches, seizures, memory loss, hallucinations, behavioral changes, and weakness of the facial nerves. Symptom onset can either be sudden or slowly progressing. Neurosarcoidosis presentation can vary widely between patients, making it difficult to diagnose. There is no cure, so symptom management and disease progression prevention are the primary treatment goals.
Optic neuritis is a condition that causes loss of vision and pain due to inflammation of the optic nerve, the nerve is responsible for relaying visual information from your eye to your brain. Although the associated loss of vision and pain is sudden, it is usually temporary, with improvements in vision occurring within weeks. Optic neuritis has been associated with several autoimmune diseases such as multiple sclerosis, infections, drug toxicity, and vitamin deficiencies. Because it is often the first symptom of a more serious disorder, it is critical for individuals who experience optic neuritis to have thorough follow-up care after initial treatment of this condition. Here at UConn Health, we have extensive experience treating and managing patients with optic neuritis and the associated conditions.
SPG11-related hereditary spastic paraplegia is a rare neurological disorder where patients start developing leg muscle weakness and stiffness. These patients have a mutation in the gene SPG11, which leads to widespread brain damage. In addition to leg weakness, patients can have cognitive impairment, difficulty controlling their bladder, and muscle wasting. SPG11-related hereditary spastic paraplegia is a progressive disease, meaning symptom severity will worsen over time. This is an autosomal recessive disease meaning that a patient needs to receive a mutated copy of SPG11 from both parents. There is no known cure for this condition, and patients require medical care to manage their symptoms and monitor disease progression.
Susac syndrome is a rare autoimmune disorder where the body attacks the small blood vessels in an individual’s ears, eyes, and brain. As a result, individuals can experience hearing loss, vision loss, headaches, memory loss, confusion, trouble walking, and slurred speech. Susac syndrome will last between 1 to 3 years, with a fluctuation of symptoms. It is critical to have appropriate medical management of symptoms to prevent long-lasting damage in individuals living with this condition. This disease most commonly affects young women aged 20 to 40 and can be devastating to both the individual and their family. Here at UConn Health, we have extensive experience treating and managing patients with Susac syndrome and the associated symptoms.
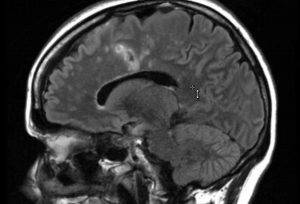
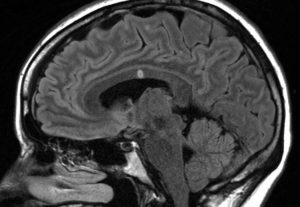
Transverse myelitis is a rare spinal cord disease that can cause varying degrees of disability. This condition causes inflammation that damages the spinal cord, disrupting pathways that relay information between the brain and the body. Individuals with transverse myelitis can experience a variety of symptoms that can include weakness, sensory alterations, and dysfunction of their autonomic nervous system, the system responsible for regulating breathing, digestion, reflexes, and more involuntary bodily functions. Although the cause of transverse myelitis is not always known, it can indicate a serious underlying disorder that might include multiple sclerosis, a rare neuro autoimmune disease, or serious infection. At UConn Health, we collaborate with experts to successfully treat and manage the immediate and long-term effects of transverse myelitis.
Call To Make An Appointment