Our research focuses on endothelial cells lining the vasculature, aiming to better understand the critical role of these cells in mediating inflammatory and immune responses. Our work has revealed post-transcriptional regulation by alternative splicing in the response to inmate immune cell recruitment.
Splicing is increasingly recognized as an important contributor to human disease. Variations resulting from changes in splicing can produce proteins with entirely new functions. Although a number of critical endothelial proteins are alternatively spliced, little is known about the regulation and function of alternative splicing in the endothelium.
We are using genetic models to examine the impact of individual splicing changes on inflammatory responses within the vasculature, and bioinformatics and high-throughput genetic screens to identify and understand the role of RNA-binding splice factors regulating these splicing changes.
We work closely with a group of talented researchers in Vascular Biology, Immunology, Neuroscience and the Institute for Systems Genomics to achieve our objectives; this includes faculty within the Center for Vascular Biology and the Calhoun Cardiology Center and the collaborators in the Immuno-Cardiovascular Group.

Projects

People

Publications

News
 Figure shows top 200 TNF induced genes in murine aortic endothelial cells with or without CRISPR-mediated deletion of Ptbp1 or a rescue with human PTBP1 cDNA.
Figure shows top 200 TNF induced genes in murine aortic endothelial cells with or without CRISPR-mediated deletion of Ptbp1 or a rescue with human PTBP1 cDNA.
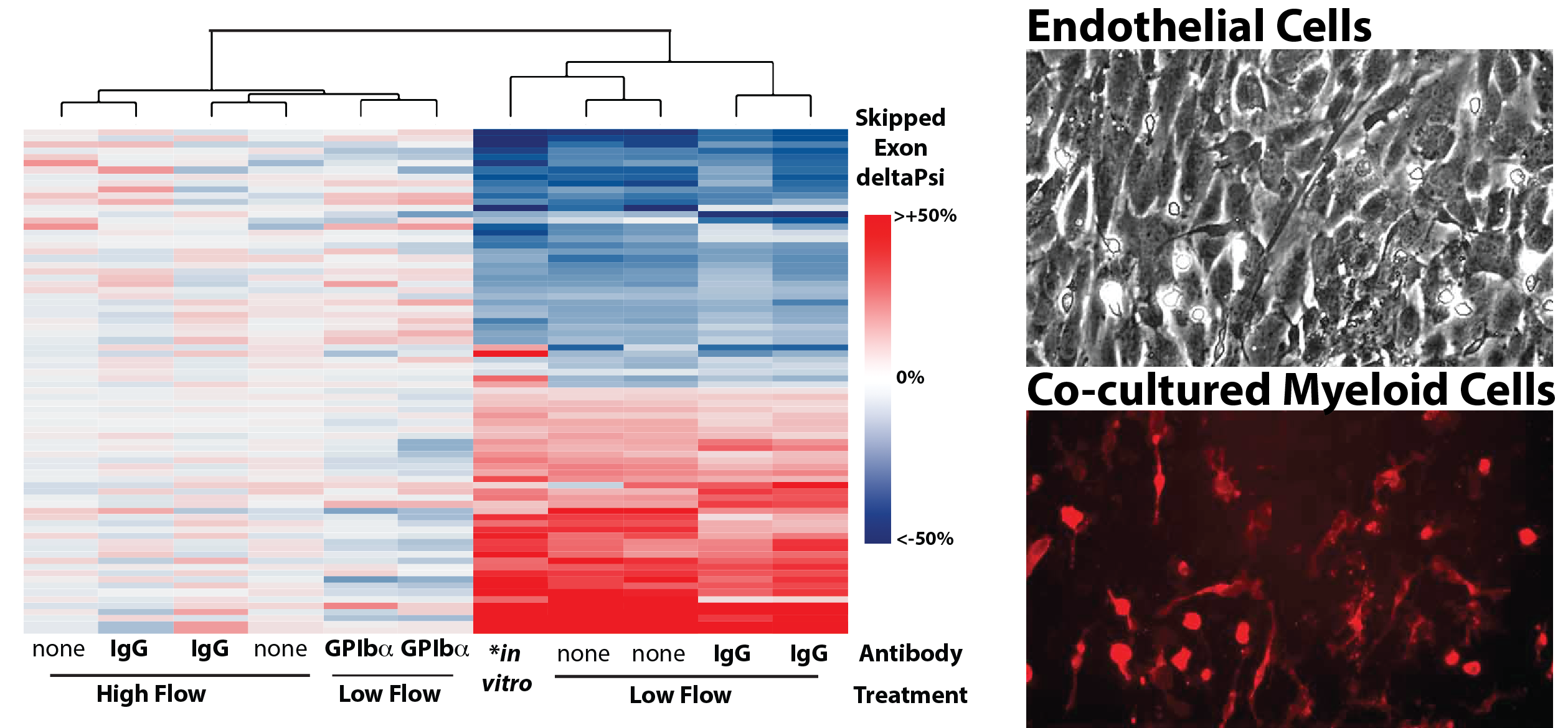
Figure shows heat map of splicing changes induced by innate immune cells in the arterial intima of the carotid artery (Murphy et al. eLife 2018). GPIba antibody was used to deplete platelets from mice prior to induction of low and disturbed flow. Note that, even in low flow conditions, platelet depletion blocks splicing changes in the endothelium. Adjacent images show, a DIC image of an aortic endothelial cell monolayer, which is overlaid with bone marrow derived monocytes from a reporter mouse (red).
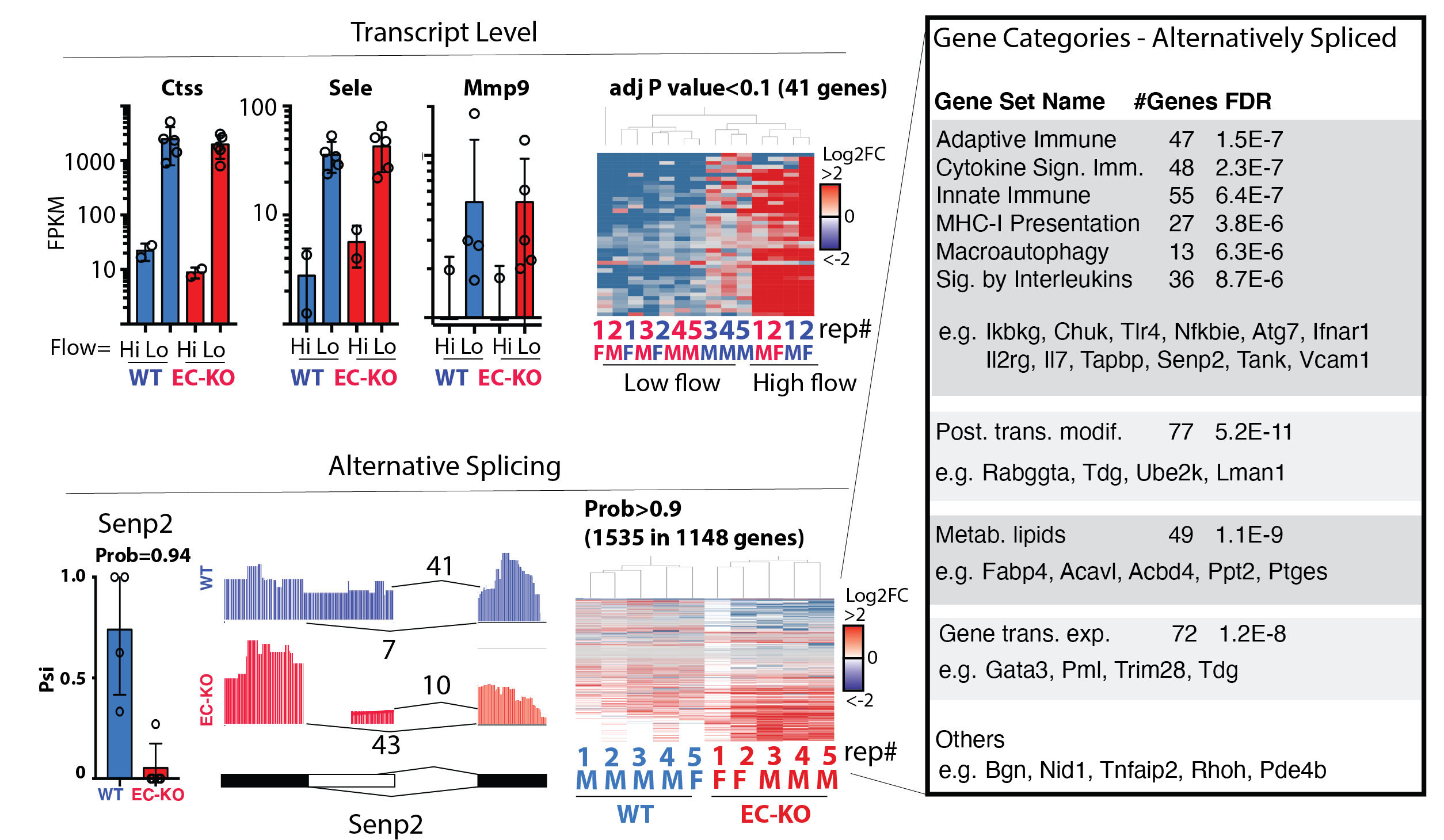
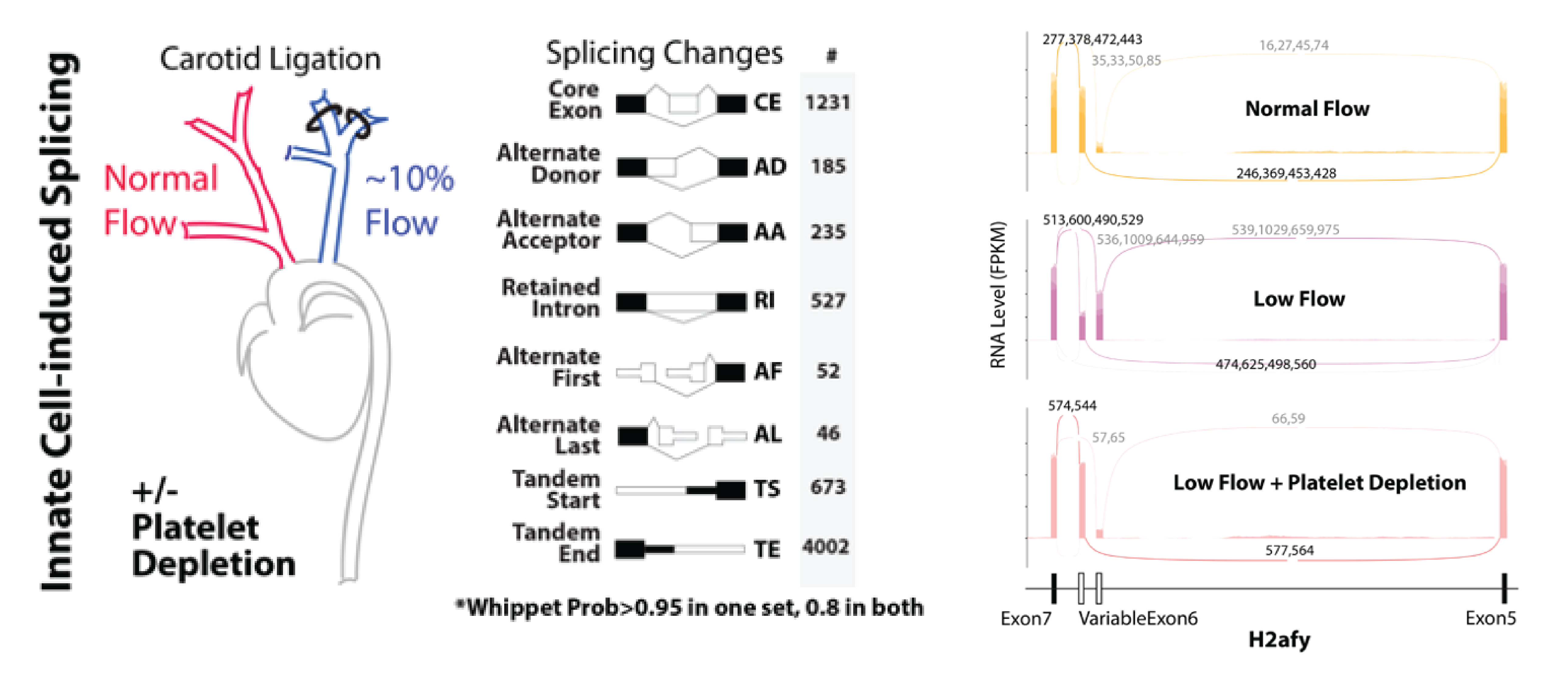
Figure shows the RNA-sequencing analysis from the carotid intima of mice exposed to disturbed flow or not. This splice factor had little effect on total RAN expression (upper panels, FPKM = Fragments Per Kilobase of transcript per Million mapped reads). However, there was a large effect on RNA splicing, primarily affected the genes and pathways in the inset.
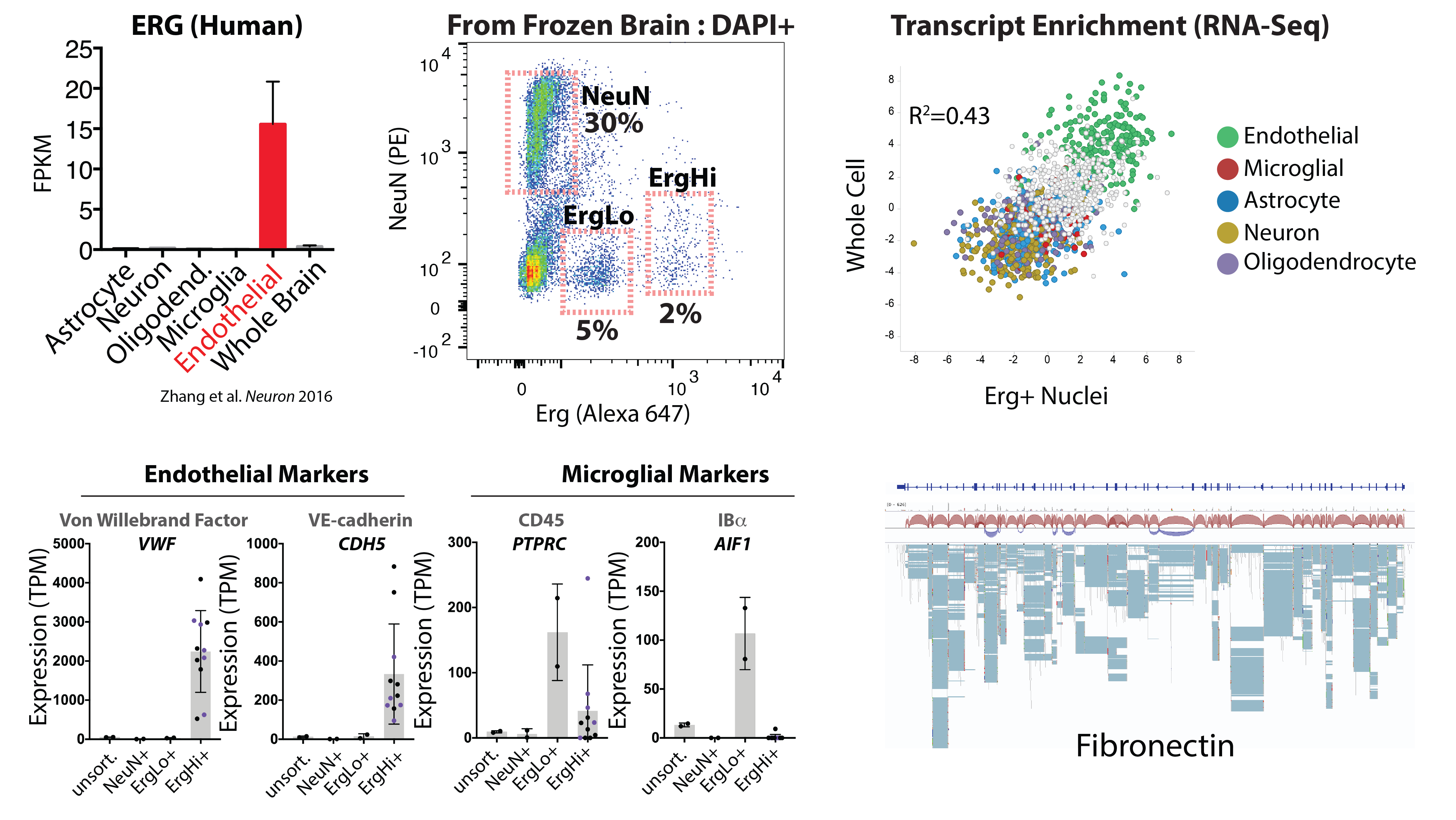
Figure cell-type specific expression of Erg in the brain in endothelial cells (from Zhang et al. 2016), and our use of an Erg specific antibody to isolate a high Erg+ population of endothelial nuclei from archived frozen brain tissues. RNA-sequencing analysis of these nuclei reveals enrichment endothelial specific transcripts (green) and a good concordance with whole brain endothelial cells. Lower panels show RNA expression levels of example markers of brain endothelial cells and microglia in the indicated sorted populations. The last panel shows analysis of the Fn transcript, and splice isoforms, in Erg+ nuclei. Details on these approaches can be found in our pre-print below.
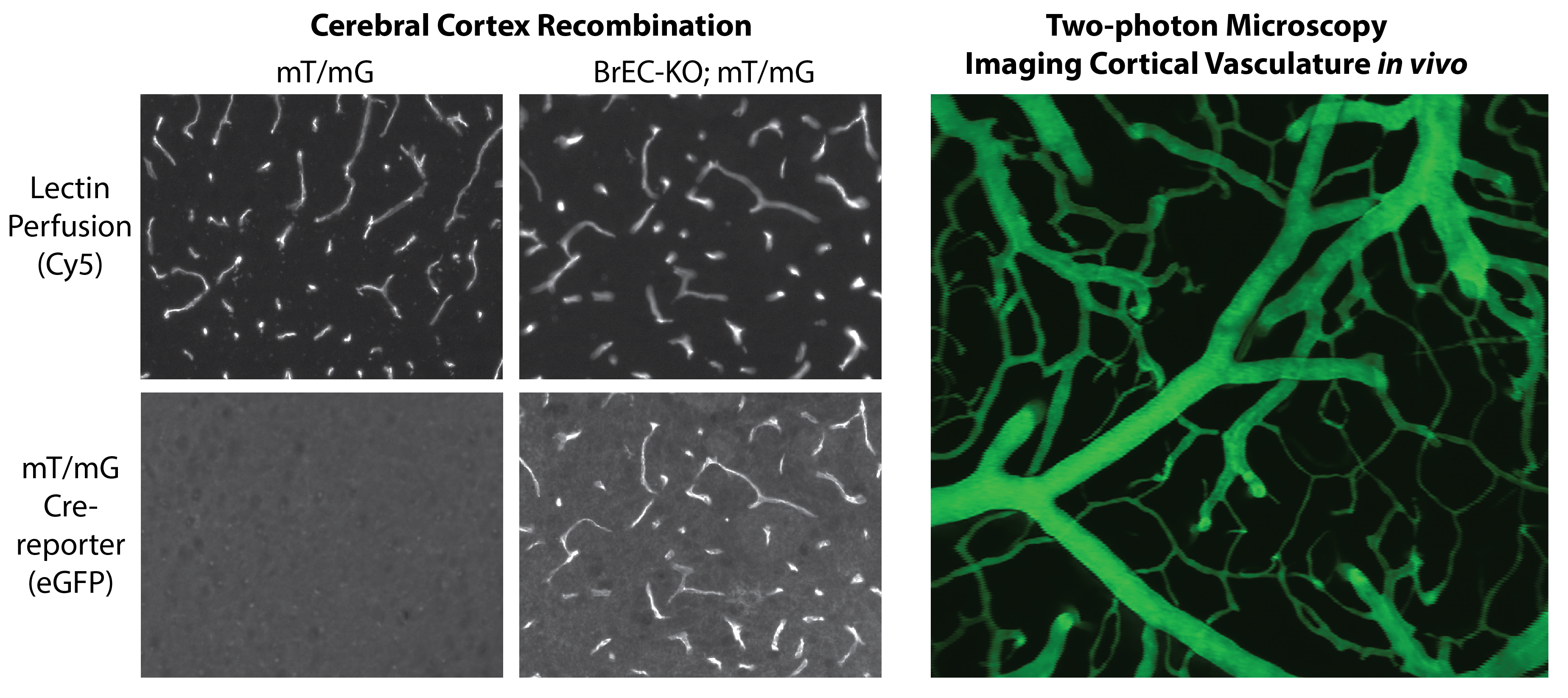
Figure shows Sclo1c1-CreER activity in brain endothelial cells, allowing the specific deletion of splice factors, such as Tardbp (TDP-43) in the brain endothelium for phenotypic characterization. Adjacent figure shows in vivo multiphoton imaging of dextran labeled vasculature through a cranial window. This approach is used to assess endothelial barrier functions, leukocyte recruitment and blood flow patterns in vivo.









September 2016 - the Murphy lab is up and running!
July 2018 - Congratulations Jess on passing your qualifying exam with flying colors.
October 2018 - Thanks to the North American Vascular Biology Organization, for recognizing Sarah-Anne's work with a Travel Award to the 2018 meeting. It was a great meeting!

October 2018 - Nice work Jess, on winning a poster prize at the 2018 North American Vascular Biology Organization meeting in Newport!
April 2019 - Congratulations Maria Xu, on your graduation from the Ph.D. phase of your program. I look forward to hearing about your next accomplishments, it was a pleasure working with you and Dr. Vella to uncover new ways in which activated T cells are retained and re-activated within atherosclerotic plaque.
May 2019 - Thank you, to the American Heart Association, for the 2019 Innovative Project Award to apply our screening approach to identify endothelial RNA-binding proteins involved in the regulation of inflammatory responses.
June 2019 - Congratulations Sarah-Anne on passing your qualifying exam, I'm looking forward to your outstanding PhD work!
September 2019 - Happy to share my thoughts on the post-doc to faculty transition through the North American Vascular Biology Organization "Lessons Learned"; NAVBO has been a key component of my success in research and I am happy to have a chance to give back!
September 2019 - We appreciate the recognition by the North American Vascular Biology Organization as "Lab of the Month".
Dec 2019 - Thank you, to the American Heart Association, for funding Sarah-Anne's graduate work on endothelial Elavl1 functions in adaptive immune responses.
Dec 2019 - Thank you, to the American Heart Association, for funding Jess's graduate work on the regulation of NFkB responses by RNA-binding proteins.
May 2020 - We were honored to be considered a finalist for the Irvine H. Page Junior Faculty Research Award from the American Heart Association, and congratulations to Hanrui Zhang of Colombia on winning the award.
May 2020 - Thank you, American Heart Association, for the Paul Dudley White International Scholar Award for the top ranked submitted abstract from a United States lab to the 2020 Vascular Discovery Meeting.
August 2020 - We are grateful for RF1 funding from the NIH's National Institute of Neurological Disorders and Stroke, to support our work on splice factor function in the endothelial cells of the blood brain barrier in Alzheimer's and related diseases. We are incredibly enthusiastic about pushing boundaries over the next five years in work with our collaborators Riqiang Yan (UCONN Health), Li Gan (Weill Cornell) and Chris Schaffer (Cornell). Watch this space!
September 2020 - excited to present three different projects at the North American Vascular Biology conference, and to hear about other exciting work on immune cell interactions with the endothelium, and endothelial contributions to atherosclerosis and neurodegenerative disease.
October 2020 - congratulations Sarah-Anne, on a great talk at the 2020 NAVBO meeting, and congratulations Jess on a great poster presentation, and a poster award!
January 2021 - we welcome Ashok as a postdoc to the lab!
February 2021 - We are grateful for R01 funding from the NIH's National Heart, Lung, and Blood Institute to support our work to examine set of RNA-binding proteins linking to transposon-derived sequence in endothelial inflammatory responses. We hope to shed light on this important group of RNA-binding proteins and their functions in physiology and disease.
May 2021 - congratulations Jess, on your graduation and new position at Vertex.
September 2021 - welcome Omar and Olivia, as new graduate students in the lab!
October 2021 - had a great time at the 2021 North American Vascular Biology Organization meeting and round-table discussion on Cell-Matrix Interactions in Vascular Remodeling with Wayne Orr, Martin Schwartz, Sophie Astrof, Brenda Lilly, Jesus Torres-Vazquez and my Chair, Linda Shapiro, and chairing exciting nano-talks session with Joyce Bischoff
July 2022 - welcome Chris, as a new graduate student in the lab!
July 2022 - had a great time at our first Gordon post-transcriptional regulation conference, and presenting work on Ptbp1 in the endothelium - will be back again!
August 2022 - Thank you, to the NIH NHGRI (and Charles Lee and Brenton Graveley, for funding Chris's graduate work on endothelial RNA-binding protein involvement in graft vs host injury!
October 2022 - The lab had a great time traveling to Cambridge to celebrate Richard Hynes Lasker award, and dinner in town
December 2022 - Thank you, to the American Heart Association, for funding Omar's graduate work on the dose dependent effects of TDP-43 loss on the brain endothelium in neurodegeneration.
March 2023 - had a great time at our first Cold Spring Harbor Brain Barriers conference - and congratulations to Ashok on his poster prize, and Omar on a great presentation!

May 2023 - welcome Stephen as a post-bac and MS student to the lab.
June 2023 - Wonderful celebration of Richard Hynes and the seminal discovery of LETS (large external transformation sensitive protein), now known as Fibronectin 50 years ago.


July 2023 - had a great time at our first Cold Spring Harbor post-transcriptional regulation conference - will be back again!
July 2023 - welcome Swati as a post-bac student to the lab, and great lab outing for drinks and yard games.

September 2023 - congratulations Sarah-Anne, on defending your thesis and your PhD - we look forward to hosting your postdoc for the completion of your work!

September 2023 - We are grateful for RF1 funding from the NIH's National Institute of Neurological Disorders and Stroke to support our work with Martin Schwartz to examine the regulation of FLT-1 splicing and contributions to Vascular contributions to cognitive impairment & dementia (VCID).
October 2023 - so much fun bringing the lab the North American Vascular Biology conference in Newport RI, and excellent oral presentations by Sarah-Anne and Omar, and poster presentation by Ashok

October 2023 - welcome Sam, as a new graduate student in the lab!
June 2024 - Congratulations Sam on passing your qualifying exam, and being award the Esperance Fellowship in Personalized Nutrition. We appreciate the support of Roger Newton ’74 MS, ’05 (HON), and the Nutritional Science Program at UConn Storrs for their support!
June 2024 - Ashok and Omar enjoyed the Gordon Brain Barriers conference in New London, congratulations on Ashok on your talk, travel award, and poster award, and to Omar on an outstanding presentation. Thank you to the Brain Barriers meeting for your support of our work!


July 2024 - Omar, Chris and Patrick enjoyed the Gordon post-transcriptional conference in Portland, an awesome meeting and great place to get new ideas!
July 2024 = Breeya, welcome to the lab as a new graduate student
August 2024 - The lab had another fun outing to the Talcott Collective, a great place for frisbee golf and some refreshments!

October 2024 - so excited for Patrick's first invited talk at the North American Vascular Biology Meeting at Asilomar in Monterey, CA. This is particularly meaningful, as Patrick has attended this meeting since graduate work with Rong Wang at UCSF in 2004. Thank you Dr. Alex Adam and Dr. Gabby Fredman for the invitation. And congratulations Omar his talk and travel award, and Ashok on his excellent presentation


October 2024 - a treat to be back on the UCSF campus to give a talk and meet with Rong Wang.

November 2024 - congratulations Omar on your accepted paper to Nature Neuroscience.


January 2025 - so nice to see Ashok representing the lab internationally, through his trip to India and talks at Indian Institute of Science Education and Research (IISER) Bhopal, Pondicherry University and Bengaluru City University. See a TV interview from his visit here.
February 2025 - congratulations Ashok, on the acceptance of your paper at Science Advances!
March 2025 - congratulations Omar, on your successful defense!
April 2025 - congratulations Ashok, on your talk, and Emily on your poster presentation. Thanks to the CSH Brain Barriers group, it was a fun meeting!


July 2025 - Swati, welcome to the lab as a new graduate student
September 2025 - Sam, congratulations on your F30 fellowship - first submission!
October 2025 - Olivia, congratulations on your selected talk at the 2025 North American Vascular Biology Organization Meeting from abstracts!
{kind=link}
November 2025 - We are honored to have been awarded seed money for an exciting project on the contributions of endothelial TDP-43 to ALS from the ALS Network. It was an amazing experience talking with the panel of scientific experts and dedicated volunteers with lived experience with the disease. We hope to make important contributions to our understanding of the endothelial contributions to disease progression.
![]()
November 2025 - Dr. Murphy is honored to receive the Marlene Cohen and Jerome Fleisch Chair in Vascular Biology at the University of Connecticut Medical School. This provides an important source of funds - prior to federal or foundational funds - to drive new project directions in the lab, enabling the next generation of exciting vascular research contributions.