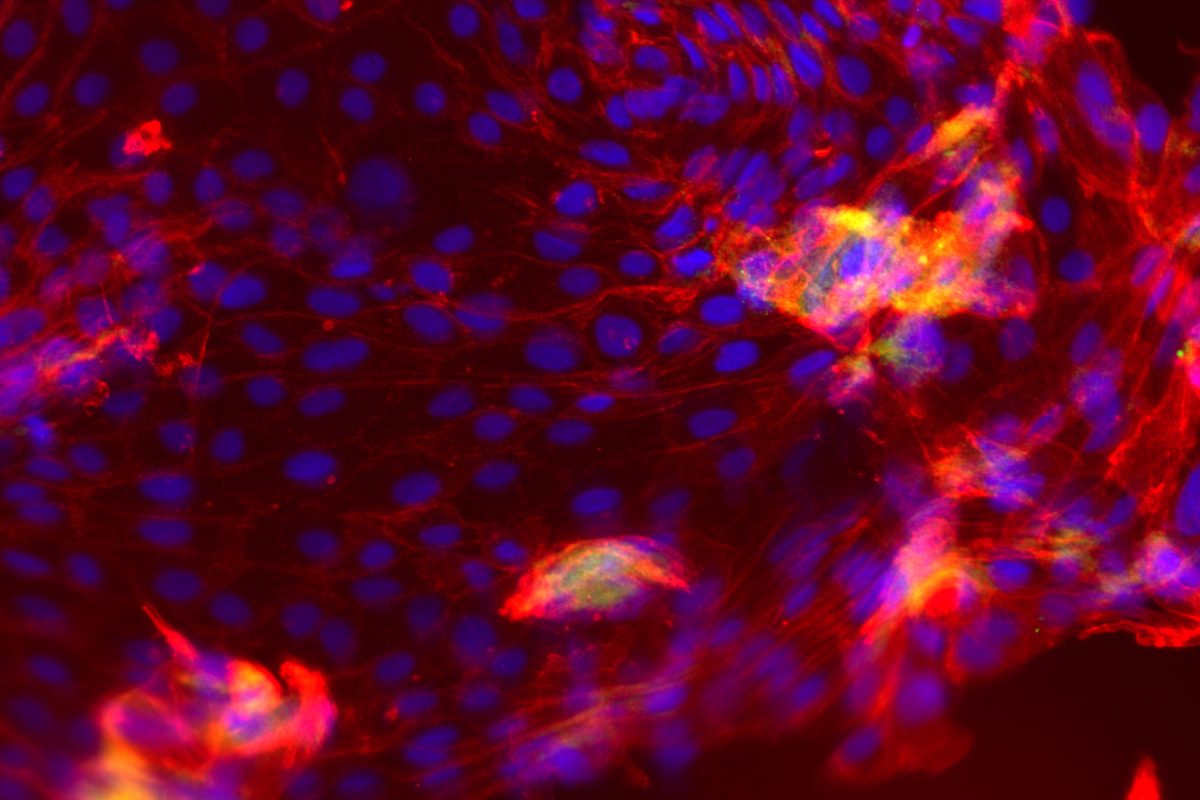
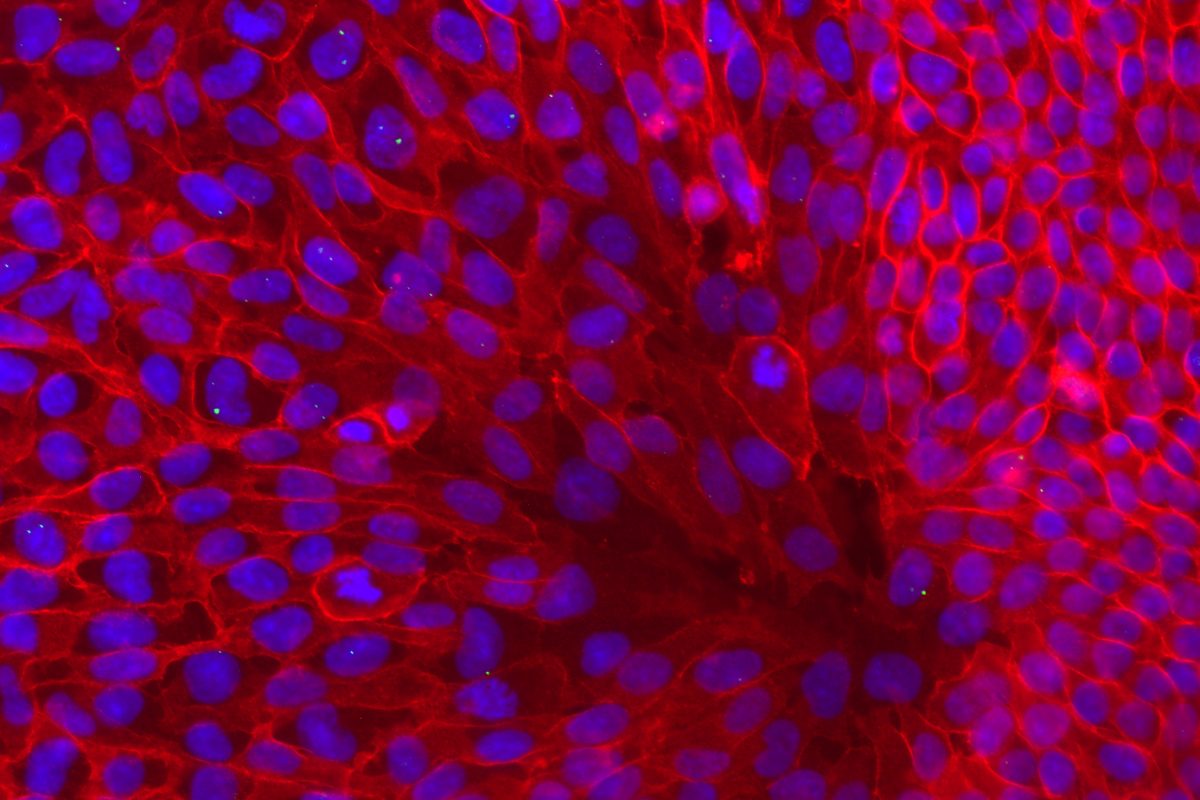
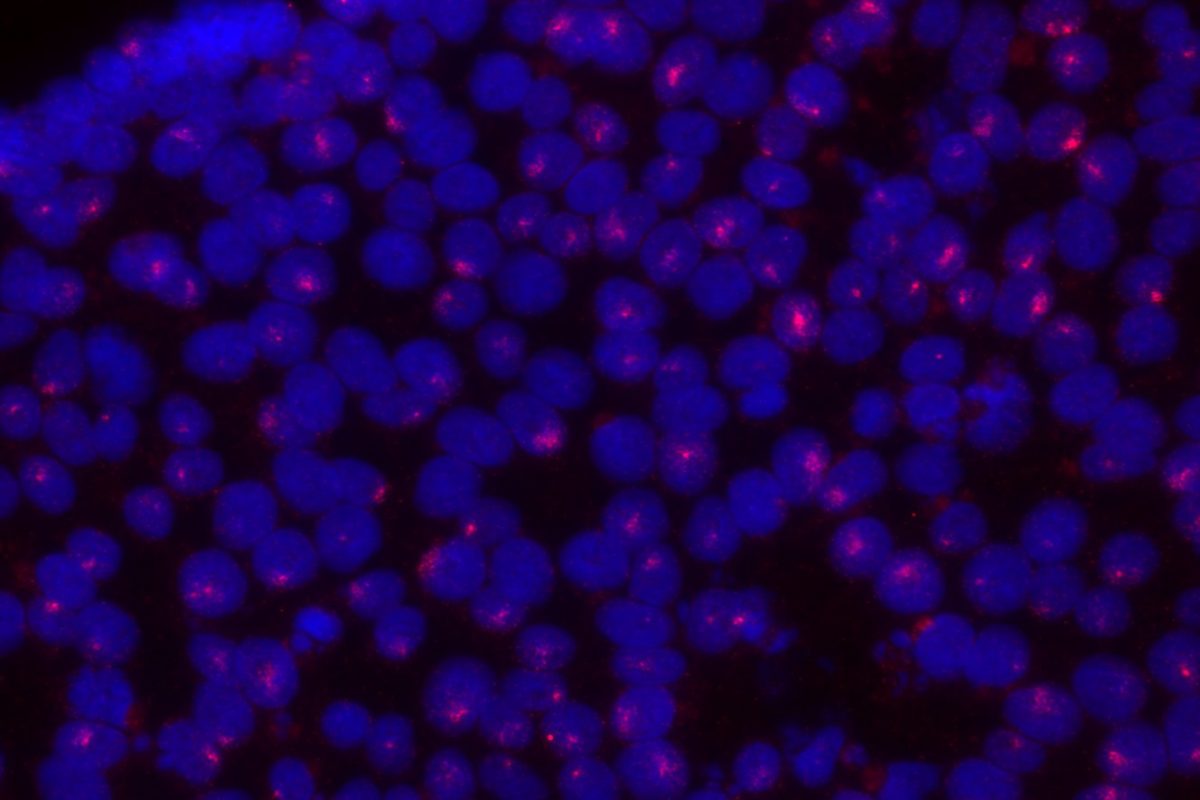
Importantly, because many escapee genes have Y-linked homologs (X-Y pair or gametologs), they are expressed from two active copies in both XY males and XX females. Turner’s syndrome (TS, karyotype 45,X) is hypothesized to result from haploinsufficiency in some of these genes, but only its cardinal phenotype (short stature) has been conclusively mapped to an X-Y pair to-date. Yet, by far the most frequent outcome of XO karyotype, is failure to reach term (est. 10-15% of all spontaneous terminations), and life-span in TS patients is reduced (~15 years) due to progressive aortic defects. We aim to use our TS hiPSC models to determine which genes contribute to the (extra)-embryonic defects that underpin these TS phenotypes.
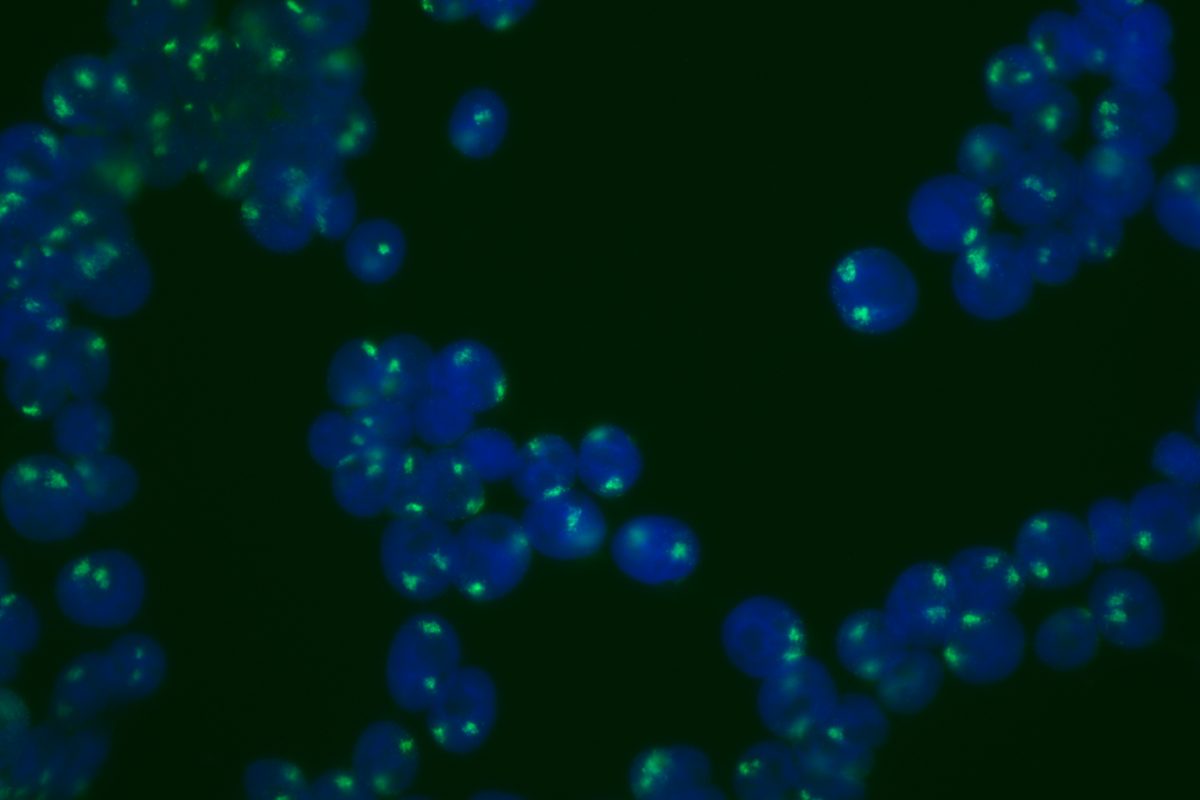
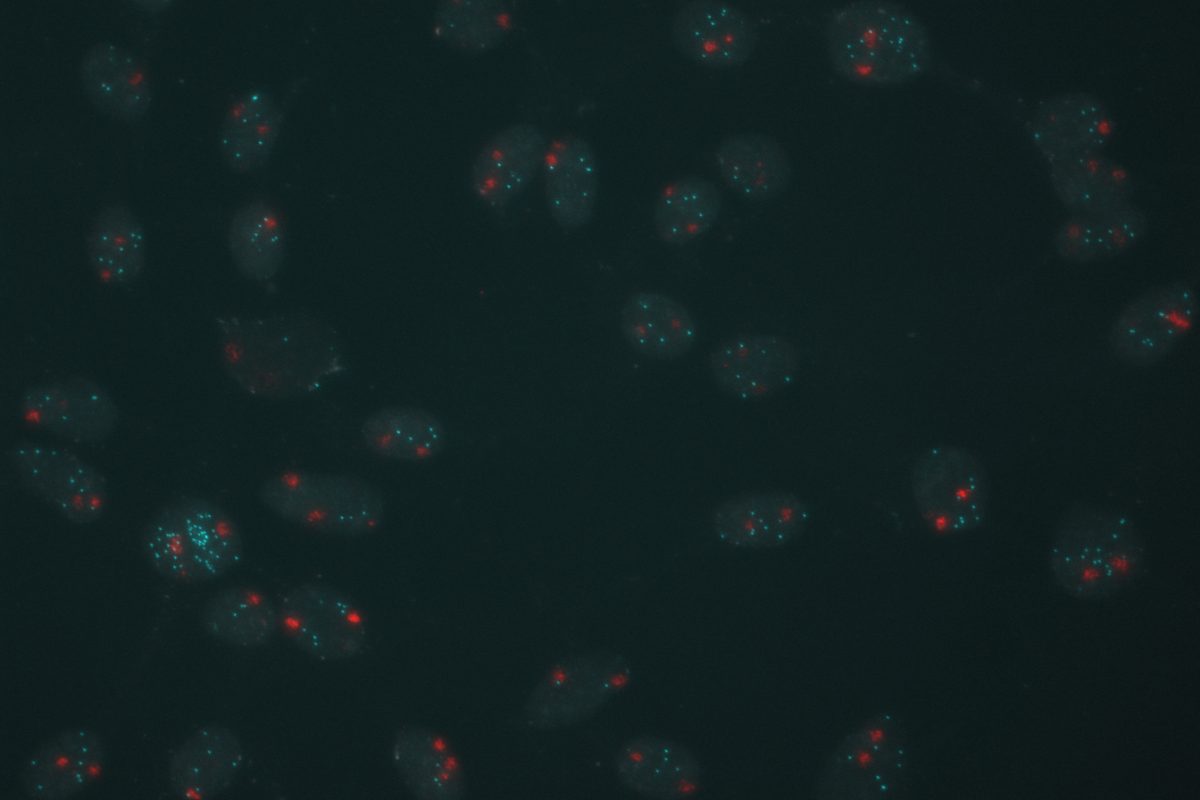
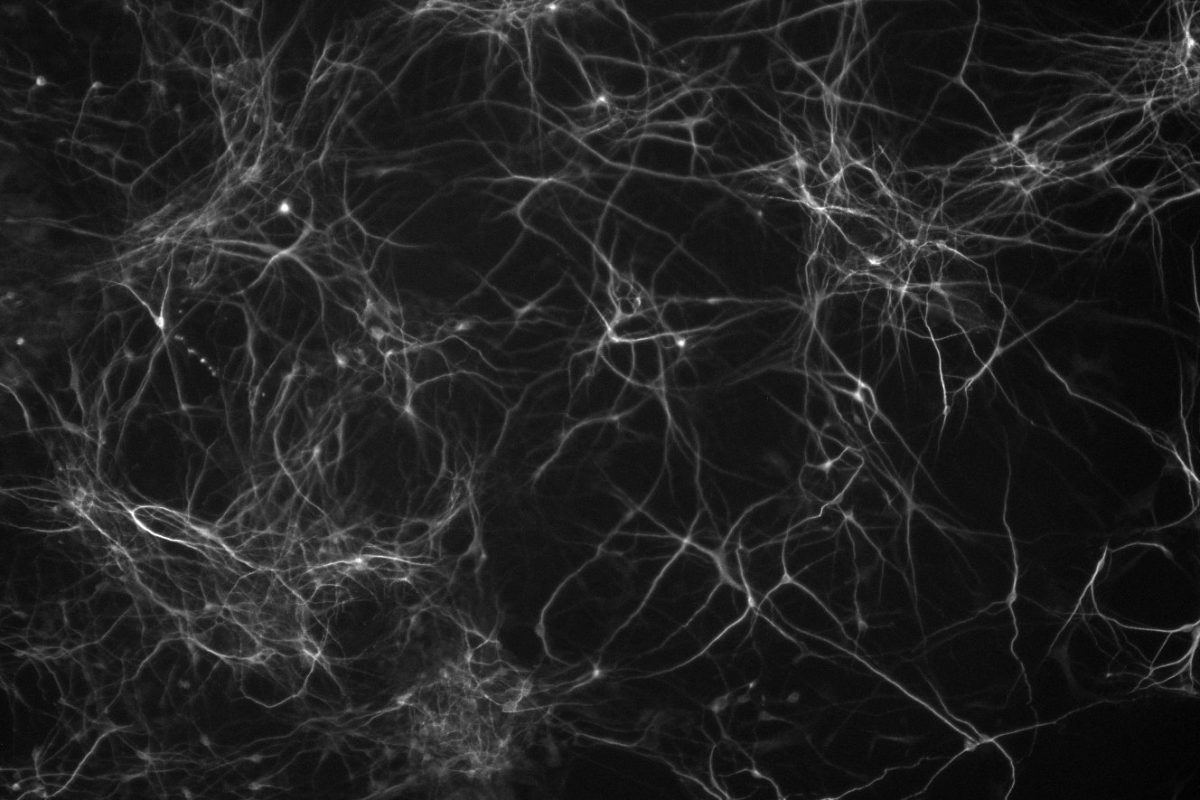
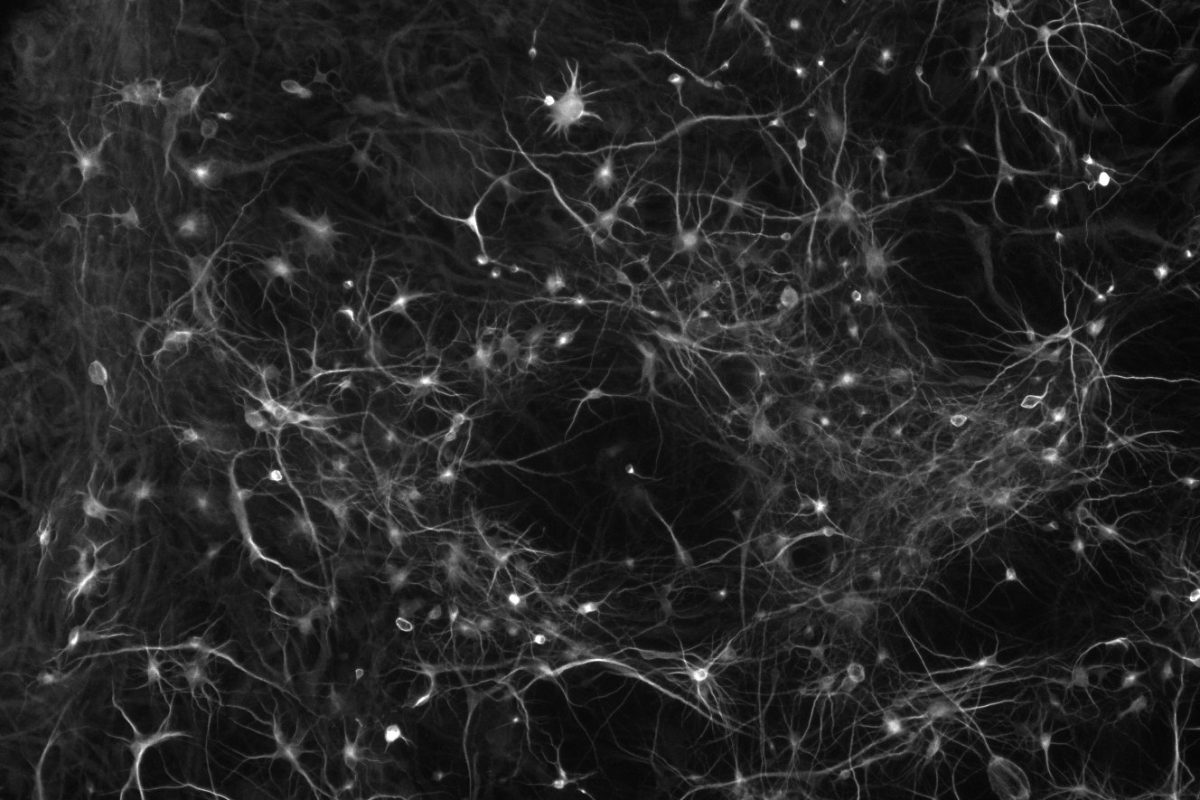
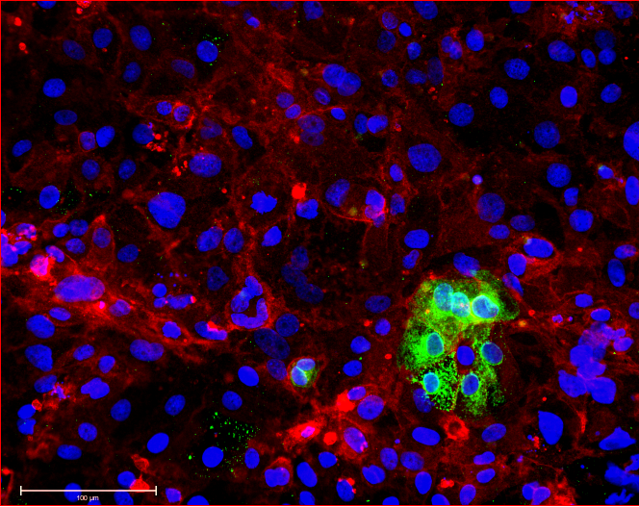
In contrast, excess gene dosage from chromosome 21 causes Down Syndrome (DS), the most frequent viable aneuploidy in humans (karyotype 47, +21 incidence of 1 in 700). DS encompasses a range of phenotypes including congenital heart disease, immunodeficiency, and intellectual disability. In concert with signs of accelerated aging, middle-aged individuals with DS tend to develop highly-penetrant, early-onset Alzheimer’s disease (AD), making DS the most frequent single genetic cause of AD overall. We are re-purposing XIST to silence one of the three copies of chromosome 21 in our DS hiPSCs learn how trisomy 21 impacts neural development. Using allele-specific CRISPR-mounted tools, we also aim to dissect which genes contribute to DS-relevant cellular phenotypes and to explore avenues to correct their dosage.